AIDD - 分子药物发现的计算方法现状总结
分子药物发现的计算方法现状总结

01
引言
药物发现的流程近年来因计算技术的飞速进步而发生了深刻变革。**计算辅助药物设计(CADD, Computer-Aided Drug Design)和人工智能驱动药物发现(AIDD, Artificial Intelligence-Driven Drug Discovery)**逐渐成为该领域的重要支柱。自20世纪80年代CADD方法的初步开发以来,它已经从最初的分子对接和虚拟筛选发展到今天结合大规模高性能计算和机器学习模型的多学科交叉领域。如今,CADD和AIDD在药物靶点发现、化合物筛选、结构优化等方面取得了显著进展,不仅提高了新药研发的效率,还降低了成本,为新药的发现与优化提供了强有力的工具。
本文将重点探讨当前小分子药物发现中的计算方法,尤其是CADD和AIDD的最新进展,分析它们的应用实例与面临的挑战,并展望这些技术未来在药物发现中的发展方向。
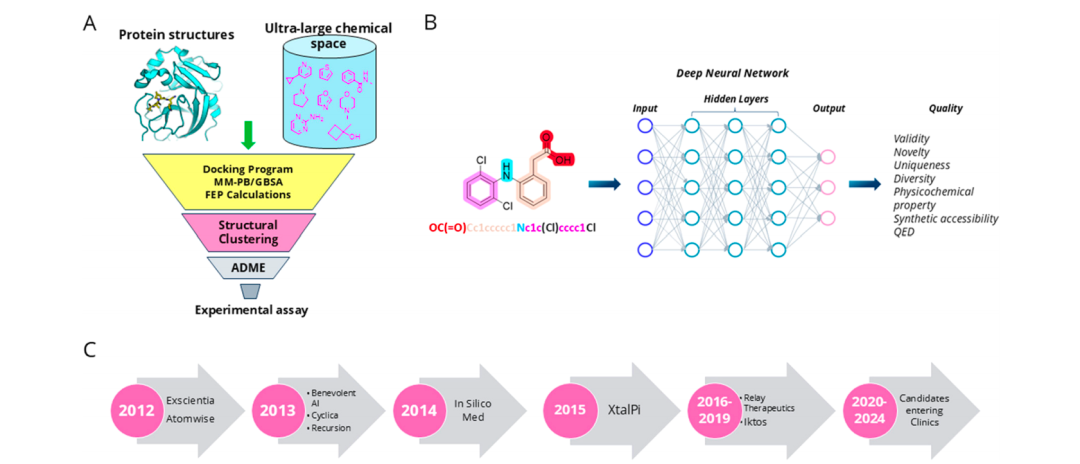
图1: (A) CADD 的一般工作流程。(B) 通过深度学习架构生成分子。© 过去十年人工智能公司发展中的例子。
02
CADD技术的最新进展
计算辅助药物设计(CADD)已经成为药物发现的主流方法之一,其核心思想是利用计算机技术和物理化学原理来模拟化合物与生物靶标的相互作用,帮助科学家筛选并优化潜在的药物候选分子。近年来,CADD技术在分子对接、虚拟筛选、分子动力学模拟和自由能计算等方面取得了显著进展。
2.1 基于结构的药物设计(SBDD)
SBDD是一种直接利用靶标三维结构信息进行药物设计的方法,主要包括分子对接、分子动力学模拟和自由能计算等技术。分子对接技术是CADD最基础的工具之一,它通过预测配体与受体蛋白的结合模式,筛选出与靶标结合能力最强的化合物。
近年来,分子对接算法的精度和速度得到了极大提升。新的分子对接软件不仅能够处理大规模虚拟化合物库,还可以结合先进的力场模型,提高结合模式预测的准确性。Lyubimov等人在多巴胺D4受体的虚拟筛选中,利用先进的分子对接算法,在短时间内筛选了超过1.38亿个化合物,并成功发现了多个新的命中化合物。
此外,**分子动力学模拟(Molecular Dynamics, MD)**作为一种模拟分子运动的技术,在药物发现中的应用也日益广泛。MD模拟通过探测配体与受体的动态相互作用,可以揭示结合机制、预测配体的结合动力学,以及发现潜在的全新结合位点。近年来,一些先进的采样技术(如增强采样技术)被引入到MD模拟中,显著提高了模拟效率和准确性,帮助科学家更好地理解配体与受体的结合行为。
**自由能计算(Free Energy Perturbation, FEP)**是一种用于精确预测配体与受体结合自由能的方法。通过结合高精度的力场模型和大量的MD模拟,FEP方法能够为药物优化提供准确的自由能信息,帮助科学家提高化合物的活性和选择性。现代的FEP方法已经得到了显著改进,能够在合理的时间内进行大规模计算,并在药物开发中得到广泛应用。
2.2 基于配体的药物设计(LBDD)
LBDD主要基于已知的配体信息,利用配体的结构与活性关系(Structure-Activity Relationship, SAR)来预测新化合物的活性。药效团模型(Pharmacophore Modeling)是LBDD中常用的工具之一,它通过提取活性化合物的共同特征,构建出能够匹配其他潜在配体的三维模型,从而筛选出具备相似活性的化合物。
此外,定量结构-活性关系(Quantitative Structure-Activity Relationship, QSAR)模型也是LBDD的重要组成部分。QSAR模型通过将化合物的分子描述符与其生物活性之间的关系进行量化,构建出能够预测新化合物活性的数据驱动模型。随着机器学习技术的发展,现代QSAR模型的预测能力得到了极大提升,尤其是在非线性数据处理和大规模数据集上的应用。
现代的LBDD技术不仅能够通过药效团和QSAR模型筛选出具有潜在活性的化合物,还能够通过构建虚拟化合物库,快速生成具有特定性质的分子结构,从而大大加速了药物发现的进程。
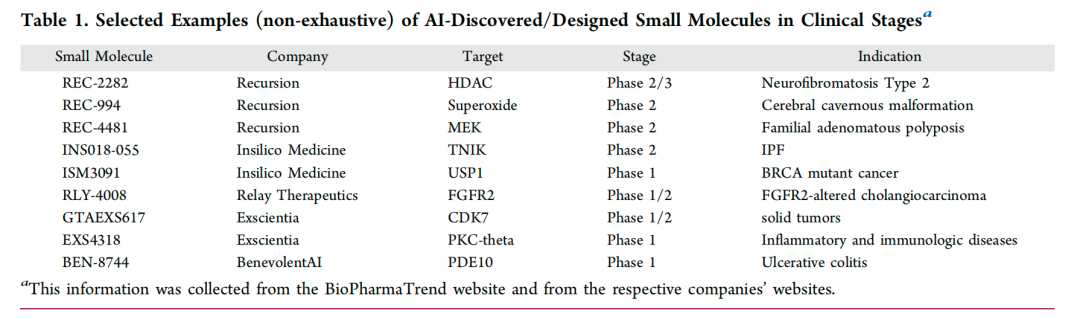
图2: 临床阶段人工智能发现/设计的小分子选定示例
03
人工智能驱动药物发现(AIDD)
近年来,人工智能(AI)技术的迅猛发展为药物发现带来了前所未有的机遇。**AI驱动的药物发现(AIDD)**结合了深度学习、机器学习、自然语言处理等先进技术,能够通过分析大量的生物数据和化合物结构,自动生成具备特定药理性质的新分子。
3.1 深度学习在药物发现中的应用
**深度学习(Deep Learning)**作为机器学习的一个分支,近年来在药物发现中得到了广泛应用。通过构建多层神经网络,深度学习能够从复杂的化合物结构和生物数据中提取特征,并生成具有特定性质的分子。
AlphaFold是深度学习在药物发现中取得的重要突破之一。该模型通过结合大规模的蛋白质结构数据和深度神经网络,显著提高了蛋白质结构预测的准确性。AlphaFold的成功不仅促进了新靶点的发现,也为基于结构的虚拟筛选提供了更加精确的结构信息。
3.2 生成对抗网络(GAN)和自编码器(VAE)
**生成对抗网络(GAN)和自编码器(VAE)**是AI生成模型中常用的两种方法,广泛应用于新分子的生成和优化。GAN通过生成器和判别器的对抗训练,能够生成具有特定化学结构和生物活性的分子。VAE则通过编码器和解码器的组合,生成潜在化学空间中的新分子结构。
例如,Insilico Medicine公司使用基于GAN的生成模型,在短短21天内设计出DDR1抑制剂,并通过体外实验验证其活性。该例子展示了生成模型在药物设计中的巨大潜力,极大缩短了传统药物发现的周期。
3.3 化学语言模型
**化学语言模型(如ChemBERT、MolGPT)**是近年来兴起的一类生成模型,借鉴了自然语言处理中的Transformer架构,通过学习分子结构的序列信息,生成具有特定药物性质的分子。与传统的化学方法相比,化学语言模型能够在更大化学空间中进行探索,并生成更加复杂的化学结构。
04
挑战与未来展望
尽管CADD和AIDD技术在药物发现中取得了显著进展,但仍然面临许多挑战。首先,AI模型的性能依赖于大量高质量的数据,而这些数据在某些特定领域可能较为稀缺。其次,尽管AI生成模型能够生成大量新分子,但其生成的分子在真实生物系统中的表现尚不完全可控。此外,如何结合CADD与AIDD技术,实现物理化学原理与数据驱动方法的完美融合,仍然是未来研究的重点。