Nature: 一种基于宏基因组序列空间生成无参考的蛋白质家族的计算方法
通过全局宏基因组学揭示功能性暗物质
Unraveling the functional dark matter through global metagenomics

Article, 2023-10-11
Nature [IF: 64.8]
DOI: https://doi.org/10.1038/s41586-023-06583-7
原文链接:https://www.nature.com/articles/s41586-023-06583-7
第一作者:Georgios A. Pavlopoulos1, 2, 3
通讯作者:Georgios A. Pavlopoulos1, 2, 3;Nikos C. Kyrpides 2
合作作者:Fotis A. Baltoumas;Sirui Liu;Oguz Selvitopi等
主要单位:
1. 希腊瓦里亚历山大-弗莱明生物医学科学研究中心生物医学基础研究所(Institute for Fundamental Biomedical Research, Biomedical Science Research Center Alexander Fleming, Vari, Greece)
2. 美国能源部联合基因组研究所,劳伦斯伯克利国家实验室 (DOE Joint Genome Institute, Lawrence Berkeley National Laboratory, Berkeley, CA, USA)
3. 希腊雅典国立和卡波迪斯特里安大学医学院新生物技术和精准医学中心(Center for New Biotechnologies and Precision Medicine, School of Medicine, National and Kapodistrian University of Athens, Athens, Greece)
- 摘 要 -
宏基因组编码的蛋白质种类繁多,反映了多种功能和活动。对这一巨大序列空间的探索仅限于与参考微生物基因组和来自这些基因组的蛋白质家族进行比较分析。在此,我们开发了一种计算方法,从宏基因组的序列空间中生成无参考的蛋白质家族,以研究目前通过参考基因组所能实现的功能多样性之外尚未开发的功能多样性。我们分析了 26,931 个宏基因组,识别出 11.7 亿个长度超过 35 个氨基酸的蛋白质序列,这些序列与 102,491 个参考基因组或 Pfam 数据库中的任何序列都不相似。利用基于大规模并行图的聚类方法,我们将这些蛋白质归入了 106,198 个超过 100 个成员的新序列群,比利用相同方法从参考基因组聚类得到的蛋白质家族数量翻了一番。我们根据这些家族的分类学、栖息地、地理分布和基因邻近分布对其进行了注释,并在有足够序列多样性的情况下预测了蛋白质的三维模型,揭示了新的结构。总之,我们的研究结果揭示了一个极其多样化的功能空间,突出了进一步探索微生物功能暗物质的重要性。
- 正 文 -
宏基因组鸟枪法测序已成为研究和分类各种生物群落微生物的首选方法。随着全基因组测序技术的最新进展以及质量和成本效率的不断提高,大规模测序变得越来越容易、快速和经济实惠。因此,在过去几年中,宏基因组测序数据大幅增加,成为研究微生物暗物质不可或缺的资源。
为阐明宏基因组样本的基因组成,通常采用两种主要方法,各有利弊。第一种方法是将测序读数精确地映射到一组已知的、有注释的参考基因组序列上,以提供已知生物、基因和潜在功能的快速概览。MG-RAST就是擅长这类分析的系统之一。在第二种方法中,将大量读数从头组装为contigs/scaffolds,可以为了解以前未被描述的生物体及其基因构成提供宝贵的信息。最近在组装和分选工具方面取得的技术进步 使得普通宏基因组的组装部分显著增加,同时宏基因组组装基因组(MAG)的数量也呈指数级增长。支持这类数据的数据管理和比较分析系统包括整合微生物基因组和微生物组(IMG/M)和 MGnify。
然而,这两种方法在基因功能注释方面都有相同的主要局限性,即依靠与参考蛋白质数据库(如 COG、Pfam 和 KEGG Orthology)的同源性搜索来预测功能。因此,在组装的宏基因组数据中预测出的基因如果没有映射到参考蛋白家族,通常就会被忽略,并从后续的比较分析中剔除。为了消除这种对参考数据集的依赖,并估算出未探索的功能多样性(被称为功能暗物质)的广度,就需要进行全对全宏基因组比较。这样的任务需要大量的计算资源,但达到这样的可扩展性水平在技术上仍具有挑战性。尽管最近报道了一些解决这一问题的杰出努力,但尚未对宏基因组进行全面调查以发现功能性暗物质。
在这里,我们提出了一种可扩展的计算方法,用于识别和描述宏基因组中的功能性暗物质。首先,我们从IMG/M的26931个宏基因组数据集中剔除了所有与IMG数据库中超过10万个参考基因组或Pfam匹配的基因,从而确定了新的蛋白质空间。接下来,我们将剩余的序列聚类为蛋白质家族,探索它们在分类学和生物群落中的分布,并在可能的情况下预测它们的三级(三维)结构。
① 新型蛋白序列空间
我们最初从所有公共参考基因组中收集了所有蛋白质序列(长度超过 35 个氨基酸残基),并在 IMG/M 平台上组装了宏基因组和宏转录组。我们总共从 89,412 个细菌基因组、9,202 个病毒基因组、3,073 个古细菌基因组和 804 个真核基因组中提取了所有蛋白质序列,最终得到了 94,672,003 条序列的数据集。本研究中的参考基因组只包括分离基因组,不包括 MAG 或单扩增基因组。同样,对于未分箱的宏基因组,我们从 26,931 个数据集(20,759 个宏基因组和 6,172 个宏转录组)(以下简称为环境数据集 (ED))中提取了所有长度至少为 35 个氨基酸且至少为 500 bp 的scaffolds预测蛋白质序列。这样就得到了一组非冗余的 8,364,611,943 个预测蛋白质或蛋白质片段。为了识别该数据集中的功能性暗物质成分,我们首先剔除了任何与 Pfam或参考基因组集中的任何序列相关的蛋白质序列。最终的非冗余目录代表了未探索的宏基因组蛋白质空间,包括 1,171,974,849 个蛋白质序列(占总数的 14%)。
② 新型蛋白家族
接下来,我们采用基于图的方法对 11 亿 ED 蛋白质进行了聚类。为了进行比较,我们对来自参考基因组的 9400 万个蛋白质采用了相同的方法。首先,通过计算所有重要的成对序列相似性,为两个基因目录(即来自参考基因组的蛋白质和来自 ED 的蛋白质)中的每一个建立了一个全对全相似性矩阵。然后对这两个图进行分析,以确定基于序列相似性的蛋白质群。为此,我们使用了 HipMCL,它是最初的 MCL 算法的大规模并行实施方案。从数据检索到聚类生成的整个过程如图 1a 所示。
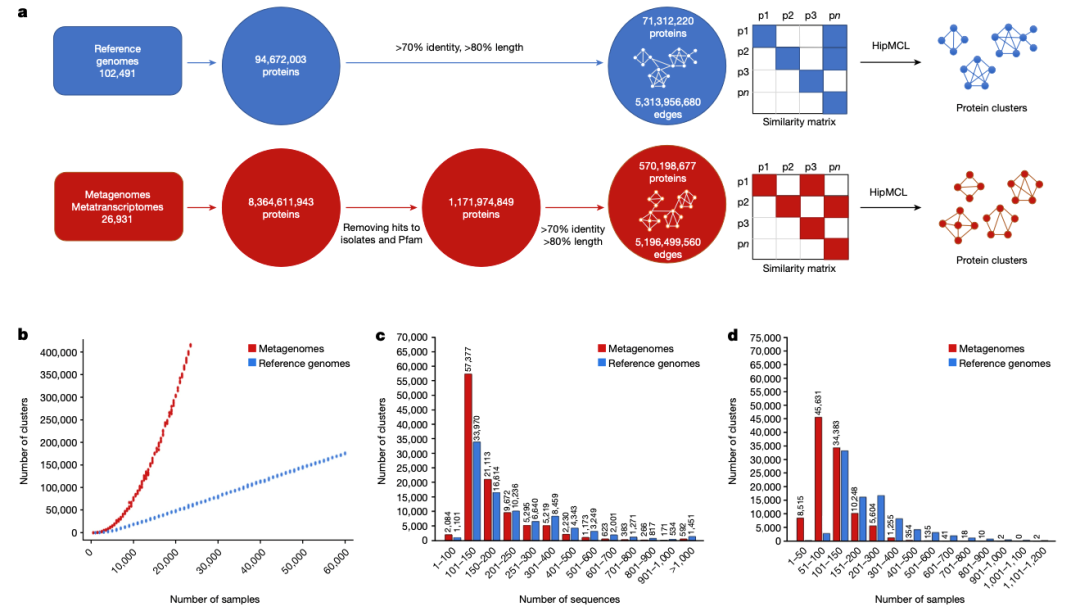
图 1:序列聚类概述。
a、对参考基因组(蓝色)和环境数据集 ED(红色)数据集中的蛋白质进行聚类。b、参考基因组(蓝色)和 ED(红色)数据集中蛋白质聚类的稀疏曲线。c、d、序列数量(c)和基因组或 ED 样本数量(d)下每个聚类的聚类成分比较和可视化。
虽然大多数至少有 50 个成员(甚至可能至少有 25 个成员)的聚类可能代表了潜在的重要功能聚类,但我们将后续分析限制在至少有 100 个成员的较大家族,以关注更高质量的数据以及预测结构的更好候选者(表 1)。我们总共发现了 106 198 个至少有 100 个成员的家族,这些家族将被称为新型宏基因组蛋白质家族(NMPFs)(表 1(右列))。为了进行比较,我们在参考基因组中至少有 100 个成员的蛋白质簇的相应集合中发现了 92,909 个蛋白质簇。通过直接比较两个聚类集(参考与 ED 蛋白质聚类集),我们发现 ED 蛋白质聚类集中至少有 3 个成员的聚类集增加了 14 倍以上,至少有 25 个成员的聚类集增加了 3 倍以上,至少有 50 和 75 个成员的聚类集增加了 2 倍左右,至少有 100 个成员的聚类集也有所增加(表 1)。虽然与参考基因组相比,宏基因组的序列空间在本质上更加破碎(补充方法和补充图 1),而且错误或不完整基因的比例会更高(这也是我们决定将所有进一步分析的重点放在较大聚类上的原因之一),但这些结果也表明,蛋白质序列空间还有很多有待探索。从≥100 个成员簇生成的稀疏曲线(图 1b)也证明了这一点。这些曲线显示,随着可用样本的增多,参考基因组的簇数呈线性增长,而宏基因组的簇数则呈指数增长,但没有达到高峰。
表1. 基于参考基因组和宏基因组的HipMCL蛋白质聚类和它们对应的不同蛋白质家族大小(累积)
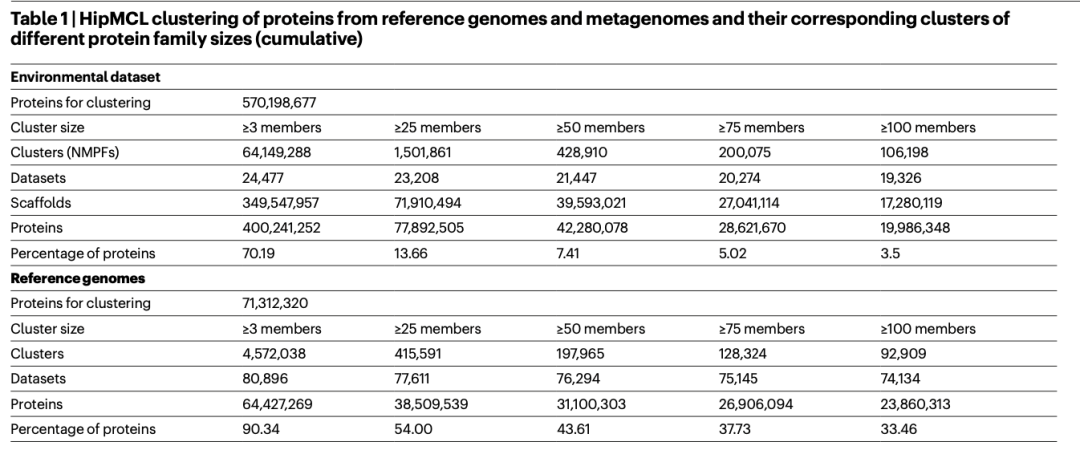
③ 生物群落分布
为了确定 NMPFs 的生物群落分布景观,我们使用 GOLD 数据库生态系统分类方案(补充表 1)收集了 IMG/M中每个样本的相应元数据。NMPF 的生物群落分布见图 2a、b 和扩展数据图 1。在这里,GOLD 三大生态系统(环境、宿主相关和工程)又被进一步划分为八种更具体的生态系统类型:淡水、海洋、土壤、植物、人类、非人类哺乳动物、其他宿主相关和工程。通过研究网络拓扑结构,我们观察到三大生态系统中每个 NMPF 内的基因共享程度最低,这与最近从 13,174 个宏基因组中观察到的蛋白质家族一致,但土壤/植物关联除外(见下文)。不过,有 7,692 个 NMPFs(7%)的成员跨越了所有八种生态系统类型。分布于所有生态系统类型的顶级 NMPFs 的属性见补充表 2,而每种不同生态系统类型的顶级 NMPFs 的属性见补充表 3 和 4。除上述分析外,还将每个生态系统进一步划分为若干子类别,以进行更精细的分析(扩展数据图 2-5 和补充图 2)。
图 2:新型宏基因组蛋白质家族(NMPFs) 的生态系统分析。
a, 八种生态系统类型中重叠的蛋白质群的UpSet图表示。不同类别之间的各种交叉用底部的图表表示,每个类别用点表示,交叉类别用直线连接。交叉集的大小用垂直条形图表示。b, 蛋白质集群及其生态系统的网络表示。根据 GOLD 生态系统分类法,应用了八种生态系统类型,由中心彩色节点(中心)表示,而灰色外围节点表示蛋白质集群。c, 八种不同生态系统类型的总 NMPFs 与特定生态系统类型 NMPFs 的分布。
土壤和植物环境中共享的 NMPFs 数量最多(占土壤相关家族的 62% 和植物相关家族的 96%),这在意料之中,因为这些生态系统的取样有很大的重叠性(即大多数植物样本来自根瘤层)(图 2a 和扩展数据图 3)。其次是土壤和淡水共有的 NMPFs,这可能主要是由于湿地和沉积物样本被归入了淡水生态系统分类。出于同样的原因,我们观察到植物和淡水 NMPF 之间以及土壤、淡水和植物 NMPF 之间有明显的重叠。相反,只有 37% 的淡水和 46% 的海洋 NMPF 相互共享。人类、非人类哺乳动物和宿主相关等生态系统类型之间共享的蛋白质家族就更少了。另一方面,人类环境和工程环境中的 NMPFs 有相当大的重叠(图 2)。考虑到工程环境主要包含来自与人类废物有关的生态系统(如固体废物和废水)的样本,这并不奇怪。同样,淡水和工程环境之间以及淡水和宿主相关类型(人类、非人类哺乳动物和其他宿主相关)之间也存在重叠,如扩展数据图 1 所示。这些重叠可能表明淡水环境受到粪便污染等现象,从而导致不同生态系统类型中同时出现相同的 NMPFs--因此也是相同的微生物群落。
在八种生态系统类型中,每种生态系统特异性 NMPFs 的百分比都有显著差异,宿主相关(非人类哺乳动物)(85.6%)和宿主相关(其他)样本(79.2%)的百分比最高,其次是海洋(48.4%),然后是土壤(14.2%)样本(图 2c)。这是因为这些生态系统类别所包含的环境具有独特的特征,例如海洋样本的海洋环境,而宿主相关类别的情况更是如此,其中包含多种具有显著生物差异的微生物组宿主(例如节肢动物和环节动物)(扩展数据图 4)。与海洋样本相比,淡水样本中生态系统类型特异性科的比例很小,这主要是由于大量湿地和沉积物样本与土壤有很强的关联性,植物/根圈相关样本与土壤样本也有很强的关联性(图 2a)。
最后,为了研究 NMPFs 在生态系统中的分布情况,对每种生态系统类型中数量最多的 NMPFs 的生态系统流行率进行了评估。每个 NMPF 在一个生态系统(例如淡水)中的流行率,是以与生态系统相关的族数据集数占研究中与生态系统相关的数据集总数的比例来计算的(补充表 3)。尽管存在与特定生态系统类型密切相关的 NMPFs(例如,超过 80% 的 NMPFs),但它们在与所述生态系统相关的总体数据集中的流行率相当低,大多数 NMPFs 分布在与每种生态系统类型相关的 5-20% 的样本中。唯一的例外是与非人类哺乳动物相关的 NMPFs,其流行率高达非人类哺乳动物数据集总数的 45%左右。
④ 分配分类
NMPFs 的分类学分配是根据 IMG 中相应scaffolds的可用分类学信息进行的,适用于簇群中的每个成员。在没有此类注释的情况下,我们结合使用了其他方法来计算推断scaffolds的分类(方法)。在包含 NMPF 成员的总共 17,280,119 个 IMG/M scaffolds中,8,049,154 个被归类为细菌,382,761 个被归类为古细菌,1,184,393 个被归类为真核细胞,1,406,588 个被归类为病毒,剩下 6,257,223 个未分类。
图 3a 和扩展数据图 6 显示了 NMPFs 根据其相应的scaffolds分类分配的分类分布情况。大多数蛋白质家族包括具有多种分类学分配(如细菌和未分类,或细菌和病毒)的序列。最大的一类是细菌/未分类序列家族,其次是病毒/未分类和细菌/病毒。被归入真核生物的科属要少得多,而被归入古细菌的科属则更少。最后,有 7,253 个聚类完全没有分类信息。
由于没有可靠的真核基因预测器可用于未分选的宏基因组 ,许多序列可能来自真核基因scaffolds,其中可能含有翻译错误(如内含子翻译错误)。然而,对这些簇的内容进行分析(补充方法)后发现,它们中的大多数除了真核生物的蛋白质外,还包括来自细菌和古细菌的蛋白质,只有极少数 NMPF 只包含真核生物序列(补充数据 6)。此外,半数以上的聚类都得到了宏转录组数据的验证。这两项观察结果证明了含有真核序列的 NMPFs 的质量。
随后,我们评估了最近从地球微生物组基因组(GEM)目录20 中鉴定出的 MAGs 中是否发现了任何 NMPF 蛋白(及其相应的家族)。具体来说,我们研究了在 GEM 目录的 52,515 个 MAGs 中,是否有任何包含 NMPFs 基因的scaffolds被分类。结果发现,只有来自 7,937 个 NMPF 的 17,953 个基因(占总数的 7.4%)(图 3b,c)出现在 GEM 目录中,其中绝大多数(93%)来自未培养物种。对于那些出现在两个或更多 MAG 中的科,我们注意到它们在分类学上的分布非常狭窄,超过三分之二的科仅限于一个种或属,只有极少数科跨越多个科、类或门(图 3d)。经统计发现,NMPFs 富集于土壤环境中常见的几个门类(例如,Gemmatimonadota、Acidobacteriota、Crenarchaeota 和 Myxococcota),而在人类和其他宿主相关环境中发现的几个门类(Firmicutes、Proteobacteria 和 Bacteroidota;图 3e)则缺乏 NMPFs。总之,这些结果表明,尽管整个测序和大规模 MAG 重建工作有所改进,但很大一部分功能多样性在分类学上仍然是孤立的。
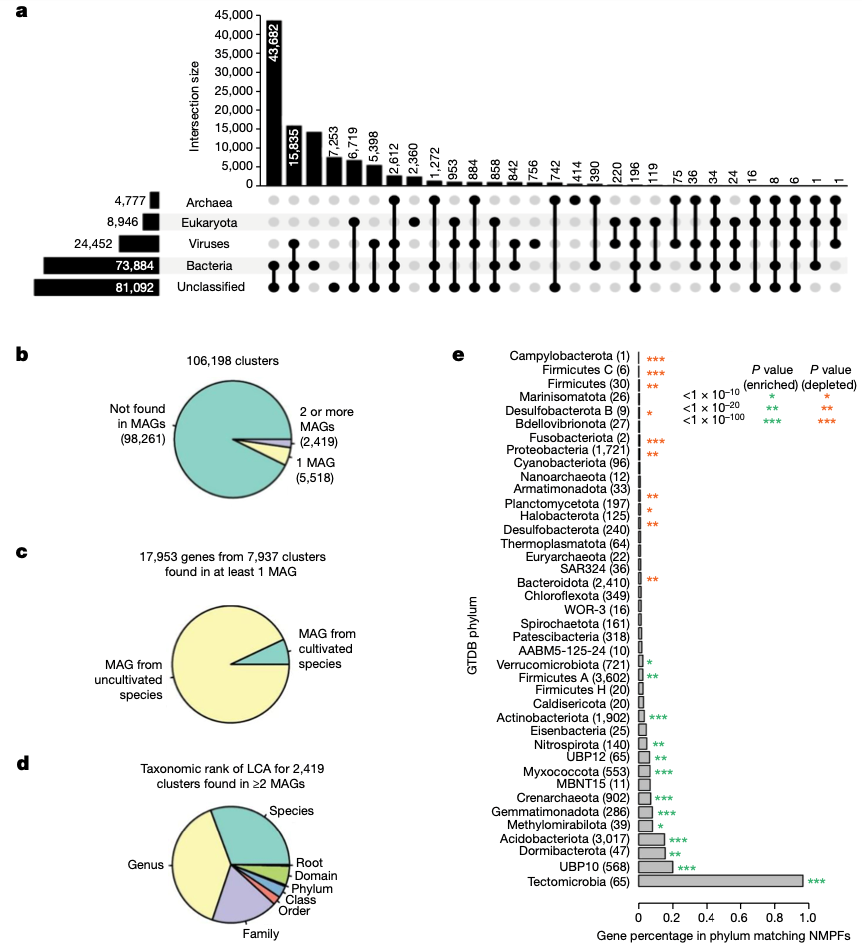
图 3:细菌和古菌 MAG 中 NMPFs 的分类组成和出现情况。
a, UpSet 图显示了新蛋白质簇的领域级分类分布。每个分类类别的总规模通过左侧的水平条形图表示。b, c, 我们确定是否在 Genomes from Earth’s Microbiomes (GEM) 目录中的scaffolds上发现了 NMPFs(b),以及是否在一个或多个栽培物种的scaffolds上发现了 NMPFs(c)。d, 在至少 2 个 MAGs 中发现的 2,419 个聚类的最低共同祖先(LCA)的分类等级。e, 与来自分配给不同门的 MAGs 的聚类相匹配的基因百分比。星号表示超几何检验的显著 P 值。绿色代表富集于该门类的基因簇;红色代表从该门类中删除的基因簇。与聚类相匹配的基因数量在门名称旁的括号中标出。
⑤ 元数据分布
我们接下来研究了 NMPF 的地理分布(扩展数据图 7)。发现有极少数科(1,372 个;1.3%)的地理分布范围有限(1 公里以内),当我们考虑到最大距离为 1,000 公里时,这一数字仅略有增加(4,330 个;4%)。这些科大多分布在植物、土壤和淡水生态系统中。这些科中有极少数包括在海洋生态系统或人类样本中发现的成员,这是因为这些生态系统中的微生物散布较广(扩展数据图 7f,g)。
大多数 NMPFs(64,186 个,占 60.44%)由来自宏基因组和宏转录本组的蛋白质混合组成,进一步证实了它们的存在,而 38,292 个 NMPFs(占 36.06%)只包含在宏基因组中发现的蛋白质,3,720 个 NMPFs(占 3.50%)只包含在宏转录本组中发现的蛋白质(补充表 5)。含有来自宏基因组和宏转录本组的成员的科的百分比随着每个科的成员数量的增加而逐渐减少。在宏基因组和宏转录组中发现的 NMPFs 也具有最广泛的样本分布,也就是说,在最多的样本中发现了这些集群(补充表 6)。这些集群大部分归类于环境生态系统(土壤,其次是海洋和淡水样本),主要包含细菌和未分类序列。
为了估计环境测序数据中新颖蛋白质群的分布情况,我们将从每个scaffolds中提取并用于本研究的新颖蛋白质数量与相应scaffolds中的基因/蛋白质总数进行了比较。大多数分析过的scaffolds(13,407,728 个或 77.59%)既包含新基因,也包含已知基因(前 20 个scaffolds;补充表 7-11)。对这些scaffolds中的新基因和基因总数进行比较后发现,scaffolds的大小或每个scaffolds的基因总数与新基因的数量无关。我们研究中最大的scaffolds(5,123,848 bp,4,302 个基因)只包含一个新序列。一般来说,研究中最大的scaffolds只包含数量有限的新序列,且来自细菌或未分类的宏基因组样本(补充表 9)。相反,含有最多新序列的scaffolds长度(和基因数量)不一,且大多来自病毒(补充表 10)。
由于大多数新型蛋白质(14,185,414 个序列)都位于已知基因的旁边,我们研究了 NMPFs 与归入同一 Pfam 家族的邻近基因的共存情况。通过将每个 NMPF 的共现 Pfam 结构域映射到其相应的 COG 功能类别,我们获得了额外的注释;这可用于提供每个家族基因邻近的进一步信息。NMPF 在不同功能类别中的分布情况见补充图 3-5。保守基因邻域表明存在功能耦合 ,因此可为推定功能预测提供更多信息。因此,在 78% 的scaffolds中发现 F004468 家族与核糖体蛋白共存(即在 151 个scaffolds中的 118 个scaffolds中编码了该家族),这表明该家族具有与翻译相关的功能。同样,在 67% 的编码scaffolds中,F021307 家族被发现位于可能的叶绿体核糖体蛋白操作子中。总共有 7625 个 NMPF 与特定 Pfam 的共存率超过 50%,而 585 个家族与一个 Pfam 家族的共存率超过 90%(补充数据 1)。这些关联还可用于预测 NMPFs 的功能作用;扩展数据图 8 和图 9 给出了几个例子,其中将选定 NMPFs 的基因邻域以关联网络的形式呈现,并结合了 COG 的功能注释。
⑥ 结构分布
最近在蛋白质结构预测方面取得的突破使蛋白质序列的结构表征变得快速而准确。宏基因组序列已被证明是发现新结构的一个特别丰富的来源。在此,我们在至少有 16 个不同序列的 NMPFs 上运行了 AlphaFold2,或在 TrRosetta预测出结构良好的蛋白质的 NMPFs 上运行了 AlphaFold2(方法)。图 4a 对结果进行了总结。在符合上述标准的 81,345 个 NMPF 中,预测出了 80,585 个三维模型,其中 13,096 个 NMPF 的预测置信度较高(预测 TM (pTM) 得分 > 0.700)。pTM 分数综合了每个位置的预测置信度和每对位置的预测配准误差(pAE),表明了结构域-结构域方向的置信度。
在结构聚类的基础上,这些高置信度预测代表了 4,361 种独特的结构。为了检验这些结构的新颖性或功能,我们将它们与 SCOP-Extended (SCOPe)26 和蛋白质数据库(PDB) 中通过实验确定的结构进行了比较。共有 3,808 个结构(12,253 个 NMPFs)与至少一个 SCOPe 结构域有明显的结构重叠(TM-score > 0.5)。其中 2,718 个结构(7,769 个 NMPFs)与至少一个 SCOPe 结构域或 PDB 组装有非难点,这表明 62.3% 的高质量预测与至少一个 SCOPe 结构域或 PDB 组装有一定的相似性。
这些基于结构相似性的新分配现在可用于相应序列的功能预测。图 4c 中显示了几个例子。例如,F034396 家族在使用 HHsearch 搜索时没有与 PDB 吻合的序列(e 值 = 12 的最高吻合),但在使用 SCOPe 结构域 d3cmba1 的结构搜索时却与 PDB 吻合(TM-score = 0.69),具有乙酰乙酸脱羧酶的功能。其他没有 HHsearch 检索结果(e 值大于 10)但结构检索结果很强的例子包括 F010804-d1z45a1(TM-score = 0.73,半乳糖突变酶)和 F097565-d1xkra_(TM-score = 0.73,趋化酶)。我们强调,这些情况应视为需要实验验证的知情预测,因为相同的折叠并不总是对应相同的功能。完整列表见补充数据 2。不过,通过将这些新分配与其他 NMPF 元数据(如基因共现)相结合,可以进行一些验证和额外的功能注释。扩展数据图 8 给出了几个例子。
为了确认其余 553 个没有 SCOPe 点击的蛋白质是新的折叠,我们对所有 PDB 生物组装进行了更彻底的搜索,包括所有可能的链排列。总共有 345 个模型与至少一个 PDB 条目有匹配,其中 305 个是额外的新分配。对剩余的 208 个模型进行了进一步过滤,删除了其中 50%的结构与 SCOPe 结构域相匹配的预测。最后,223 个 NMPF 中的 162 个折叠和/或结构域-结构域方向被确定为新的(图 4b)。这些折叠的完整列表见补充数据 3。
虽然由于缺乏任何重要的结构同源性,无法对这些新折叠进行可靠的功能注释,但可以从它们的相关元数据中获得一些有关其潜在功能的提示。扩展数据图 9 举例说明了三个具有新型结构褶皱的 NMPF 的基因邻域和生态系统元数据。
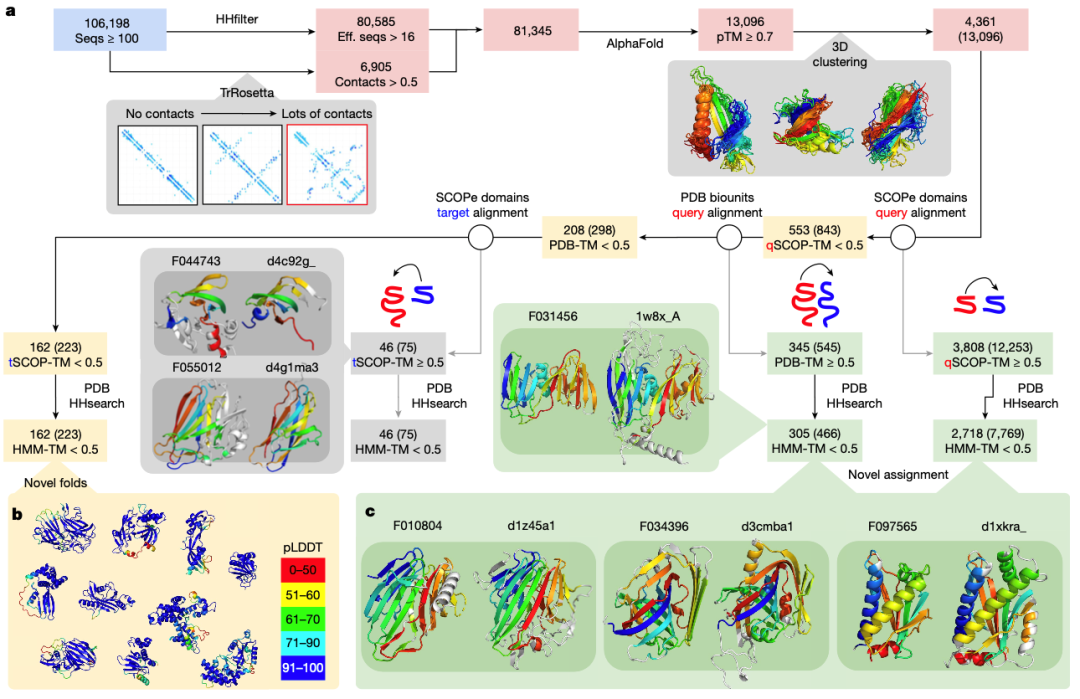
图 4:NMPF 的结构特征。
a, 向 AlphaFold 提交了至少有 16 个有效序列(eff. seqs)或许多接触的蛋白质簇。过滤结果包括预测置信度高(pTM ≥ 0.70)的结构,然后根据成对 TM 分数计算对这些结构进行聚类。工作流程的所有后续步骤都显示了独特聚类的数量,并在括号中显示了 NMPF 的总数。由于过滤是在 NMPF 层面进行的,因此只有括号内的数字才是总和,因为同一聚类的成员有可能在每个 TM 分数过滤步骤中处于不同的位置。每个预测结构都与 SCOPe 域进行了比对。对 SCOPe 没有命中的模型进行进一步比对,如果有任何命中的完整 PDB 集合或其中一个 SCOPe 结构域与至少 50%的预测结构比对,则对其进行筛选。使用 HHsearch 对照 PDB 进一步筛选结构域(来自 SCOPe 匹配结果)或多结构域(来自 PDB 匹配结果)。b, 与 SCOPe 或 PDB 都没有显著匹配的模型被认为是潜在的新折叠。c, 与 SCOPe 结构域或 PDB 生物组装体有匹配但没有显著 HHsearch 匹配(HMM-TM-score < 0.5)的模型被认为是新分配。
- 讨 论 -
可以认为,估计和探索微生物功能多样性的最佳方法是对序列多样性空间进行系统编目和详尽描述。在过去的三十年中,对成千上万培养微生物菌株的基因组测序使这一序列空间得到了前所未有的发展和表征,揭示了以最大限度地提高系统发育多样性为目标的测序工作可以进一步发现和发展目前已知的蛋白质家族多样性。尽管对相应序列编码功能多样性的探索远远落后,但已发现的新蛋白家族数量的激增在很大程度上伴随着对其中一些家族进行有针对性的功能表征的增加,特别是在重要的生物技术应用领域,如发现新的 CRISPR-Cas 基因和系统。宏基因组学的出现通过发掘隐藏的未开发序列信息宝库,进一步推动了发现新酶活性的热潮。然而,除了首次生成重要的特定生境环境基因目录之外,对呈指数级增长的宏基因组序列空间的大多数探索都集中在扩大先前已知蛋白质家族的多样性和表征上。
为了缓解这一局限性,并在全球范围内深入了解新序列空间的范围,进而了解整个测序生物群落的功能多样性,我们收集了近 27,000 个公开的宏基因组和宏转录组数据集。从这些数据集中,我们生成了 NMPF 目录,其中包括 106,198 个宏基因组蛋白质家族,这些家族有 100 个或更多成员,与参考微生物基因组或 Pfam 条目中的基因没有序列相似性。虽然这些家族的数量与从所有生命领域的 10 多万个整合到 IMG 中的参考基因组中生成的家族数量相比仅仅是重复,但在含有 25、50 或 75 个以上成员的家族中却观察到了更大的增长,这有力地表明了广泛的序列和功能多样性仍未得到开发。我们预计,随着对更多新环境样本进行测序,未开发微生物蛋白质空间的这种多样性将在未来几年继续增加。
虽然本次分析所获得的宏转录组数量要少得多(4739 个;17.6%),但我们观察到大多数 NMPFs(60%)由在宏基因组和宏转录组中发现的基因编码的蛋白质组成,这表明这些基因大多表达活跃,进一步支持了这些聚类的有效性。此外,92%的聚类有跨越 50 个或更多样本的成员,而 50%的聚类来自分布在 100 个或更多样本中的蛋白质(图 2d),这也证明了聚类的质量。
在最近重建的 GEM 目录 MAGs 上鉴定出 7.5% 的 NMPFs 表明,随着我们不断获取未培养微生物多样性的遗传内容,越来越多的分类学上无主的新蛋白家族将得到分类学上的分配,这是对其功能和生态特性进行鉴定的重要一步。
本研究使用的宏基因组数据和方法存在一些局限性。需要考虑的一个限制因素是,本研究中使用的大多数scaffolds都很短(短于 5 kb)。不过,需要注意的是,由于要求比对覆盖率至少达到 80%,潜在的截短序列必须足够完整,才能与全长序列(定义为位于较长scaffolds的中间部分)聚类。这一要求在很大程度上排除了用片段蛋白质富集 NMPF 的可能性。不过,即使是含有大量此类可疑序列的 NMPFs,也能生成稳定的三维模型(通常具有较高的结构质量,pLDDT 和 pTM 分数就是证明),其中许多与 SCOPe 结构域具有结构同源关系。因此,包含此类序列的族可能代表构成多域序列的蛋白质片段或蛋白质结构域,或多聚体复合物中的组分。另外一个潜在的限制因素是序列数据集中包含了真核序列,这可能会在分析中引入误差。然而,如图所示(分类分布;补充方法),真核生物scaffolds的贡献相对较小,而且大多数相关的 NMPF 在序列比对中也包含了来自宏转录组和/或原核生物分类群的数据,这支持了它们的有效性。不过,在用于宏基因组的可靠的真核基因预测器问世之前,真核基因以及未分类的 NMPF 和序列都应谨慎处理。
- 方 法 -
① 数据收集与筛选
所有公开的宏基因组、宏转录组和参考基因组都是从 IMG/M 数据库(数据库发布时间:2019 年 7 月)中获取的。使用 tantan 应用程序删除了低复杂度区域。我们总共从 89,412 个细菌基因组、9,202 个病毒基因组、3,073 个古细菌基因组和 804 个真核基因组中提取了所有蛋白质序列。这相当于 87,084,214 个细菌蛋白、221,027 个病毒蛋白、2,464,569 个古细菌蛋白和 4,902,193 个真核生物非冗余蛋白,最终得到 94,672,003 个序列的数据集。使用 hmmsearch 工具(HMMER v.3.1 软件包)和默认可信截止值检测 Pfam 命中(v.31)。使用 LAST35 计算与参考基因组中蛋白质的匹配。我们认为,在 70% 的长度范围内(查询和研究对象之间的双向),任何一致性大于 30% 的比对序列都是命中序列。序列选择、过滤和分析程序的详细工作流程见补充方法。补充数据 4 提供了每个宏基因组和宏转录组数据集所含序列的完整摘要,包括与 Pfam 和参考基因组的比对、用于聚类的序列(见下文)以及剩余的未注释序列。
② 序列聚类和分析
序列聚类采用 HipMCL 算法,以同一性得分作为输入,膨胀参数为 2.0。之所以选择 HipMCL 而不是其他聚类解决方案,是因为它具有可扩展性和并行化能力,而且能够高效地对超大数据集进行聚类(补充方法)。聚类前,使用 LAST(70% 序列同一性,80% 比对覆盖率)计算了所有与所有的配对比对。参考基因组图由 71,312,220 个节点(蛋白质)和 5,313,956,680 条边(成对相似性)组成。ED 蛋白质图由 570,198,677 个节点和 5,196,499,560 条边组成。值得注意的是,在构建相似性矩阵的过程中,94,672,003 个参考蛋白质中的 23,359,783 个(约占 24.67%)和 1,171,974,849 个 ED 蛋白质中的 601,776,172 个(约占 51.34%)仍然是单体。使用 NERSC Cori 超级计算机(英特尔 KNL 分区)的 2,500 个计算节点(170,000 个计算核心),HipMCL 在 24 分钟内完成了参考蛋白质图的聚类,在 3 小时 20 分钟内完成了 ED 蛋白质图的聚类。
值得注意的是,为了完成这项任务,我们探索了几种基于图和序列的方法,如 CD-HIT、UCLUST、kClust或 Louvain,但这些方法都无法达到这种规模,MMSeq2-linclust40 和 SPICi41 也无法达到这种规模。补充方法》中对这些不同聚类策略进行了比较。关于序列同一性,我们将截止值设定为 70%,以实现灵敏度和特异性之间的折中。虽然其他资源使用了更严格的参数(如 MGnify,其序列聚类的序列同一性截止值为 90%),但我们选择了一种基于灵敏度的方法,但又不过分灵敏,以避免噪声、多域效应和假阳性产生的假象。在比对覆盖率方面,我们选择了高覆盖率阈值(80%),以确保生成更高质量的序列比对,并避免潜在的假象,如涉及明显截断/不完整基因、假基因和嵌合序列的部分命中。通过考虑具有 100 个或更多成员的 MCL 聚类,最终数据集的整体质量得到进一步提高,从而确保排除虚假的基因产物。补充方法》中还分析了聚类截止值(特别是序列同一性)如何影响聚类结果的质量。
对于每个聚类,使用 Clustal Omega 计算多序列比对(MSA)。然后使用 Python 编写的脚本以及 ProDy/Evol 和 Biopython 模块(90% 的序列同一性,75% 的比对覆盖率)对得到的 MSA 进行过滤,以生成种子(非冗余)比对(补充方法)。随后的所有分析步骤,包括长度分布计算、HMM 图谱训练和三维结构预测,均使用生成的种子 MSA 进行。HMM 图谱使用 HMMER v.3.1 套件的 hmmalign 工具生成。使用 Biopython(补充方法)计算每个聚类的代表性共识序列。使用 HMMER(纳入阈值:7.0 total,5.0 domain)对照整个 Pfam-A8 数据库(v.33)HMM 配置文件搜索共识序列,并使用 BLAST(30%同一性,50%双向覆盖)对照参考基因组搜索共识序列。对≥100 个成员的初始 NMPF(来自 19,473 个数据集的 113,752 个簇,包含 20,211,137 个scaffolds和 21,260,914 个蛋白质)进行了处理,以更严格的标准进行额外过滤,根据计算出的共识轮廓序列,剔除与 Pfam-A 模型或参考基因组中基因有微弱相似性的序列。这样就得到了一组更为严格的 106,198 个聚类。发现共识序列与 Pfam-A 或参考基因组有匹配的聚类都被删除了。每个 ED 数据集的聚类过程的完整摘要见补充数据 3。图谱分布使用 R 和 R/ggplot2 软件包计算。在所有关于 NMPF 序列长度的计算中,都计算并使用了每个聚类的种子 MSA 的平均序列长度。
③ 验证蛋白质家族的新颖性
使用 LAST(矩阵:BLOSUM,间隙打开:11,间隙扩展:2)和 HMMER(包含阈值:7.0 total,5.0 domain),分别使用 NMPF 簇的共识序列和 HMM 剖面,并应用 70% 的比对覆盖率截止值,对 RefSeq 的参考蛋白质组(2021 年 11 月发布)进行了额外的搜索。这些搜索结果表明,有 5,111 个聚类(约占总数的 5%)在 134,273 个 RefSeq 序列中获得了阳性命中。这些序列的分类注释显示,75,215 条(56.01%)阳性序列为真核生物,46,793 条(34.85%)为细菌,9,296 条(6.92%)为古细菌,2,905 条(2.16%)为病毒,还有 64 条序列没有分类注释。约有一半(70554 条或 52.54%)的命中序列是 2020 年以后发表的,其中大部分与 MAGs 中的基因匹配。将 RefSeq 检索结果与 UniProt(2021_04 版)记录进行交叉检验显示,31242 条 RefSeq 序列(23.27%)与 33628 个 UniProtKB 条目(453 个 SwissProt 和 33175 个 TrEMBL)进行了映射;这些序列仅对应于 32 个 NMPF 簇。其余的 RefSeq 序列(103,031 条)包含在 UniProt 的 UniParc 档案中,没有注释证据(手动或自动生成)。NMPF 聚类对照 Pfam-B 进行了搜索,Pfam-B 是一个未注释的、计算生成的数据集,包含 Pfam-A 未涵盖的序列的排列。结果,有 8,313 个独特的簇与 5,310 个 Pfam-B 有正向匹配。最后,对每个数据集搜索的阳性命中进行了汇编和比较。总共有 12,846 个聚类在 RefSeq、Pfam-B 或两者中都有阳性命中,而其余的聚类(93,352 个)则没有命中。最后,根据 AntiFam v.6.0 对所有 NMPF 序列进行了搜索,AntiFam46 v.6.0 是一个 HMM 配置文件集,旨在检测潜在的虚假蛋白质序列、伪基因和错误蛋白质翻译。只有 43 个序列被识别出来,其中有两个 AntiFam 图谱的命中率和比对覆盖率较低(<50%)。
④ 蛋白质簇的生态系统和分类注释
通过相关的环境数据集,用可用的环境和分类元数据对 NMPF 聚类进行注释。在环境元数据方面,使用 GOLD生态系统分类方案 将数据集组织成生态系统组(如淡水、海洋、土壤、宿主相关);然后根据 IMG 中发现蛋白质序列的 ED 样本的生态系统信息,将每个蛋白质集群分配到一个或多个生态系统中。除 GOLD 外,环境本体(ENVO)和地球微生物组项目本体(EMPO)也被视为潜在的替代分类系统。使用与每个 ED 样品相关的 GOLD 生物样本项目元数据,将 ED 样品和 NMPF 映射到 ENVO 和 EMPO。所有三种分类系统的生态系统分配见补充数据 5。最终采用了 GOLD 分类法,因为它提供了最多样化的选择,并对所有 ED 样品和 NMPF 进行了分类。在包含 NMPF 的 19,326 份环境样本中,14,540 份(75.24%)是环境样本(如土壤、淡水),3,867 份(20.01%)是宿主相关样本(如人类、植物),919 份(4.76%)来自工程环境(如废水、工业废物)。
以类似的方式,利用每个IMG/M数据集所含scaffolds的NCBI分类信息(如有),对聚类进行了初步分类注释。需要注意的是,本研究中使用的大部分scaffolds都太短,因此仍未进行分类。此外,有关病毒脚手架分类学的信息也非常少。为了缓解这些问题,我们使用了之前已被确定为病毒并包含在 IMG/VR 3.0 版本中的大于 5 kb 的scaffolds注释。此外,使用 DeepVirFinder(v.1.0) 对长度为 1-5 kb 的scaffolds进行分析,随后使用 R 软件包 qvalue51 将生成的 P 值转换为 q 值,以获得假发现率的估计值。q≤ 0.001 的scaffolds被保留为推定病毒scaffolds。使用两个真核序列检测工具 Whokaryote 和 EukRep 对未分类的scaffolds进行进一步分析。此外,还根据 Tara Oceans 收集的真核生物 MAGs 对 NMPF 簇进行了搜索。最后,使用 MMseqs2 分类工具对所有剩余的未分类scaffolds进行分类学分配,根据 UniRef 进行六帧翻译搜索,并将每个分析scaffolds分配给每帧最佳命中的最低共同祖先。NMPF 簇的分类分配基于源scaffolds,见补充数据 6。关于分类注释和分析的详细描述见补充方法。
通过 Gephi 使用 Yifan Hu 算法 生成网络布局并将其可视化,对生态系统和 NCBI 分类群中的蛋白质集群进行了分布分析。由于考虑到所有聚类,生成的网络非常密集,因此使用关联阈值对数据进行过滤以提高清晰度,只保留每个聚类中至少有 2% 的成员被分配到某个生态系统或系统发育的边缘。此外,还使用 R/chorddiag 和 Processing/P5 库分别绘制了环状图和分布矩阵,以进行其他分析。此外,还使用 R/ggplot2 和 R/plotly 库在 R 中创建了条形图。利用自然地球公共领域资源库(https://www.naturalearthdata.com/)中的地图制作了地理分布可视化图。
⑤ 序列质量控制
通过考虑源 ED scaffolds的预测基因坐标和基因密度,对分析中使用的预测蛋白质序列的质量进行了评估。特别是对 IMG/M 管道确定为真核生物的scaffolds,以及没有分类注释和密度较低的scaffolds进行了评估,因为后者通常表示潜在的真核生物等位基因或具有替代遗传密码的等位基因。此外,还评估了每个 NMPF 序列与各自scaffolds末端的距离,以检测可能缩短/截断的基因。根据上述方法,建立了一系列指标来评估 NMPF 聚类的质量。分析详情见补充方法。NMPF 的质量评估见补充数据 7。
⑥ 蛋白质聚类与 Pfams 的共现
NMPFs 与已知蛋白质结构域的共存性是通过在包含新的和已知蛋白质编码基因的分析scaffolds中搜索是否存在 Pfam 蛋白质结构域来确定的。使用 HMMER 和 HMM 配置文件的默认可信截止值,根据 Pfam 的 HMM 配置文件搜索每个scaffolds中已知基因的翻译序列。所有阳性命中都被归入各自的scaffolds,反过来,又被归入包含这些scaffolds中的新序列的 NMPF,作为潜在的共现域。计算每个 Pfam 域在每个 NMPF 中的共现频率百分比,即含有该域的scaffolds数量占与 NMPF 相关的scaffolds总数的百分比。随后将 Pfam 结构域映射到 COG 结构域及其功能类别。在 7,885 个 NMPF 中没有观察到与 Pfam 的关联;对于其余的集群,根据其频率得出的前五个 Pfam 和 COG 命中率见补充数据 1。此外,利用 NORMA(v.2.0) 和 COG 功能分类提供的注释,所选 NMPFs 的基因邻域被可视化为关联网络。
⑦ 蛋白质折叠预测
多序列比对
多序列比对的查询序列由种子多序列比对的中心序列或枢轴序列决定。在每个 MSA 中定义查询序列的方法是进行成对距离计算,创建一个全对全距离矩阵,然后选择汉明距离最小的序列。然后以中心序列为指导重新计算 MSA,过滤去除与查询序列对齐度较差的序列(序列同一性的截止值设定为 90%,对齐覆盖率的截止值设定为 80%),以及对齐度较差的位置(列占据率低)。如果最终的 MSA 有 16 个以上的有效序列,则考虑对其进行进一步分析。计算使用 Python 和 TensorFlow2、SciKit 和 Biopython 库。
用于初步筛选的 TrRosetta
初步筛选时,使用 TrRosetta 对每个推定蛋白质家族进行分析,以获得馏分图。值得注意的是,距离图是一个张量,包含每对残基的预测距离分布。通过对小于 8 Å 的距离求和,馏分图可转换为接触图,显示接触的概率。最高概率的平均值已被证明与结构准确性高度相关 ,可用于筛选可能结构良好的蛋白质。这一指标的计算速度非常快,使我们能够快速扫描 106,198 个示例。除了具有足够有效序列的 MSA 外,我们还选择了平均概率至少为 0.5 的 MSA 进行 AlphaFold 预测。
用于最终预测的 AlphaFold
由于没有一个 NMPF 与已知的蛋白质家族(Pfam)或 PDB 中的结构相匹配,因此在没有模板输入的情况下运行 AlphaFold2。每次运行生成五个模型,并选择具有最佳 pLDDT 平均值的模型进行下游评估。
搜索结构同源物
在进行结构搜索之前,先删除预测置信度较低(pLDDT < 0.7)的区域。为了测试与实验确定的结构是否存在任何结构相似性,对 SCOPe(2021 年 10 月 21 日,v.2.0.8)中的每个结构域进行了 TMalign 搜索,SCOPe 是 SCOP 蛋白结构域数据库的注释版本。为测试命中(TM-score > 0.5)是否为新分配(不易从遥远的序列同源物推断),还对 HHsearch 命中最高的结构计算了 TMalign 分数。最后,由于目标物和查询物之间的长度差异,TM-score 可能较低。这可能是由于查询是多域的缘故;为了考虑到这一点,我们使用 MMalign66 对整个 PDB28(2021 年 12 月 17 日访问)进行了额外搜索。为了进一步确认 PDB 中的任何命中都是非琐碎的,我们使用 HHsearch67 将预测结构与 HMM-HMM 对齐搜索中命中率最高的 PDB 结构进行了比较。在 HHsearch 结果支持下,与 SCOPe 有非微小匹配的预测结构被称为新分配结构。
聚类
为了进行聚类,我们对 pTM > 0.7 的所有预测结构计算了全对全 TMalign 分数,并将其修剪为 pLDDT > 0.7 的区域。聚类被定义为一个网络的连接部分,其边缘由目标对查询和查询对目标配准的 TM-score > 0.6 所定义。
参考文献:
Pavlopoulos, G.A., Baltoumas, F.A., Liu, S. et al. Unraveling the functional dark matter through global metagenomics. Nature 622, 594–602 (2023). https://doi.org/10.1038/s41586-023-06583-7
- 作者简介 -

希腊瓦里亚历山大-弗莱明生物医学科学研究中心生物医学基础研究所
Georgios A. Pavlopoulos
副教授
第一作者兼通讯(第一):Georgios A. Pavlopoulos
希腊瓦里亚历山大-弗莱明生物医学科学研究中心生物医学基础研究所副教授,生物信息学主任。主要从事生物信息学,基因组学,生物网络,可视化,文字挖掘等领域。曾在美国能源部联合基因组研究所从事博士后工作(2015-2018),导师为Nikos C. Kyrpides。在Nature, Science, Nature Biotechnology等期刊共发表94篇论文, h指数33。

美国能源部联合基因组研究所,劳伦斯伯克利国家实验室
Nikos C. Kyrpides
微生物组数据科学组负责人
通讯作者(第二): Nikos C. Kyrpides
美国能源部联合基因组研究所,劳伦斯伯克利国家实验室微生物组数据科学组负责人。主要从事微生物组研究,研究重点是微生物组数据科学,实验室开发了微生物基因组和微生物组比较分析平台IMG/M。担任PeerJ, Environmental Microbiology,Microbiome,Life杂志编委,在Nature, Science, Nature Biotechnology,PNAS, NAR等期刊共发表700多篇论文,h指数119,被评选为2020年汤姆森路透社高被引/最具影响力科学家。